Welcome to PyMethylProcess’s documentation!¶
https://github.com/Christensen-Lab-Dartmouth/PyMethylProcess
To get started, download pymethylprocess using Docker (joshualevy44/pymethylprocess) or PIP (pymethylprocess) and run pymethyl-install_r_dependencies.
There is both an API and CLI available for use. Examples for CLI usage can be found in ./example_scripts.
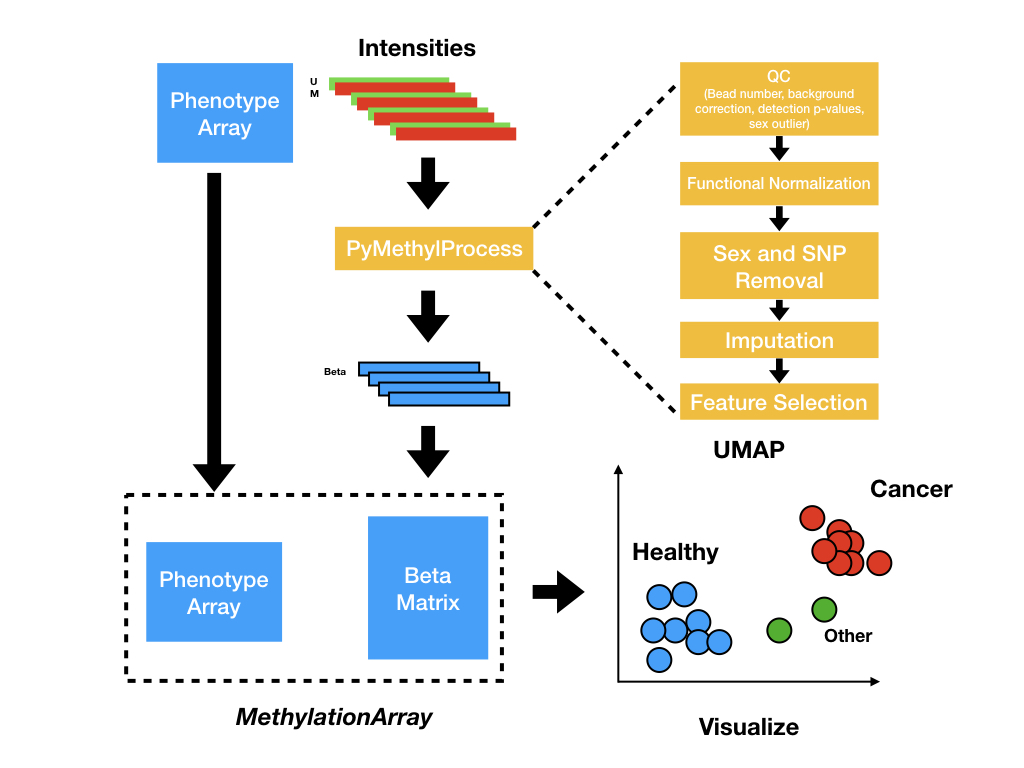
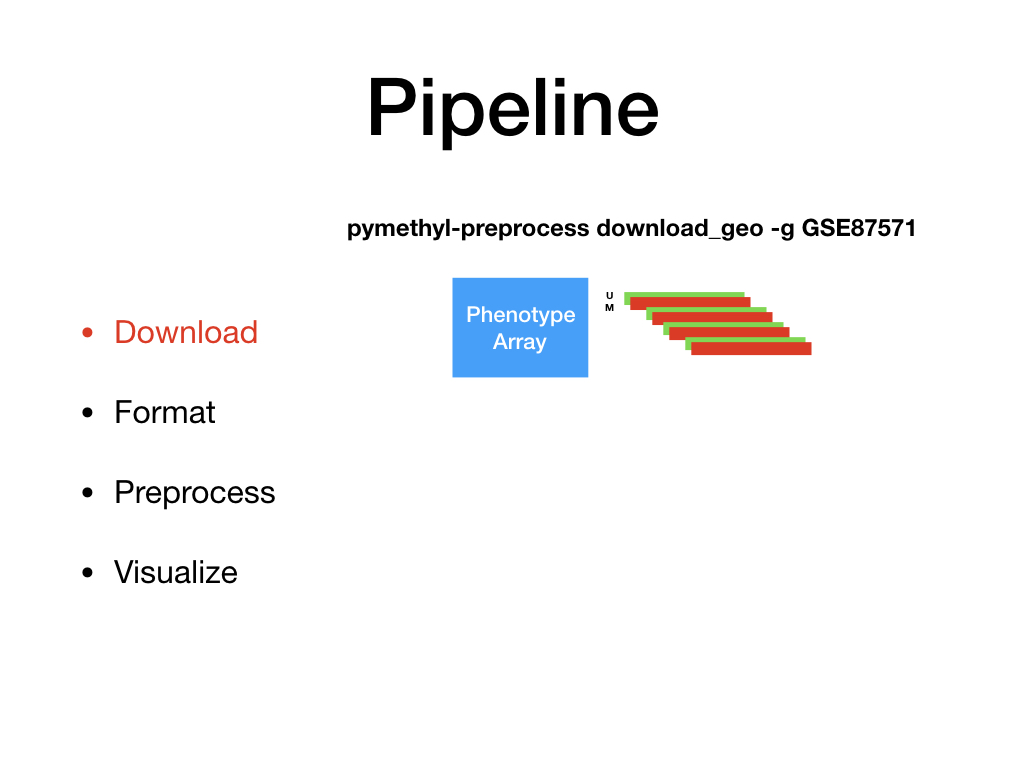
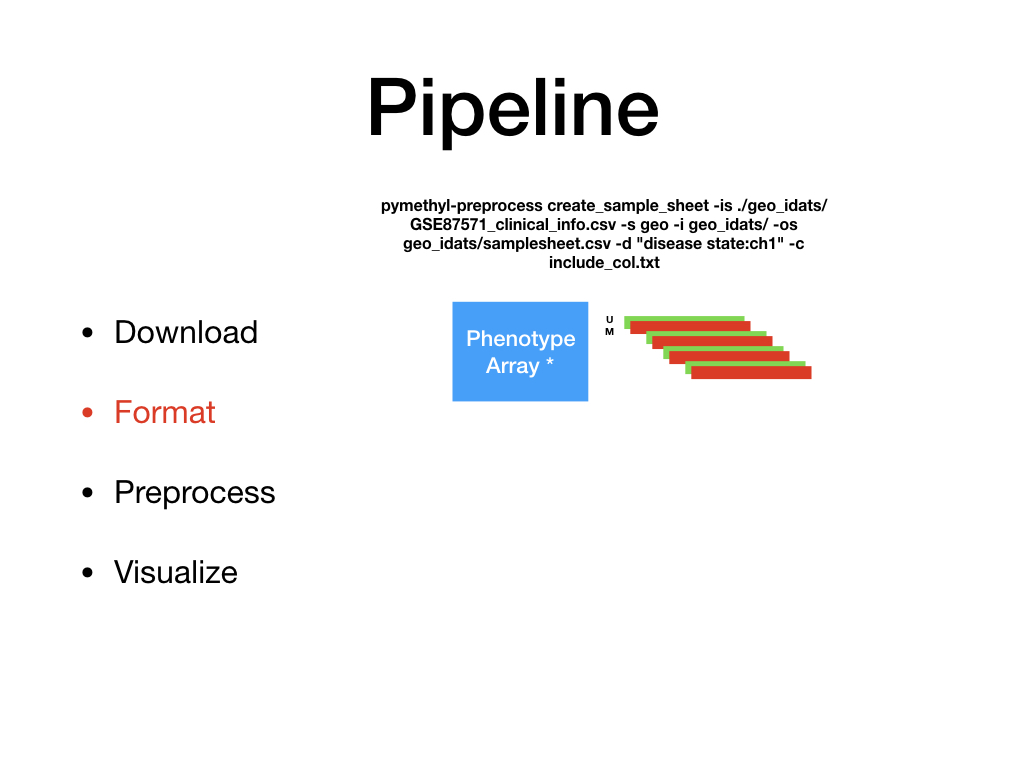
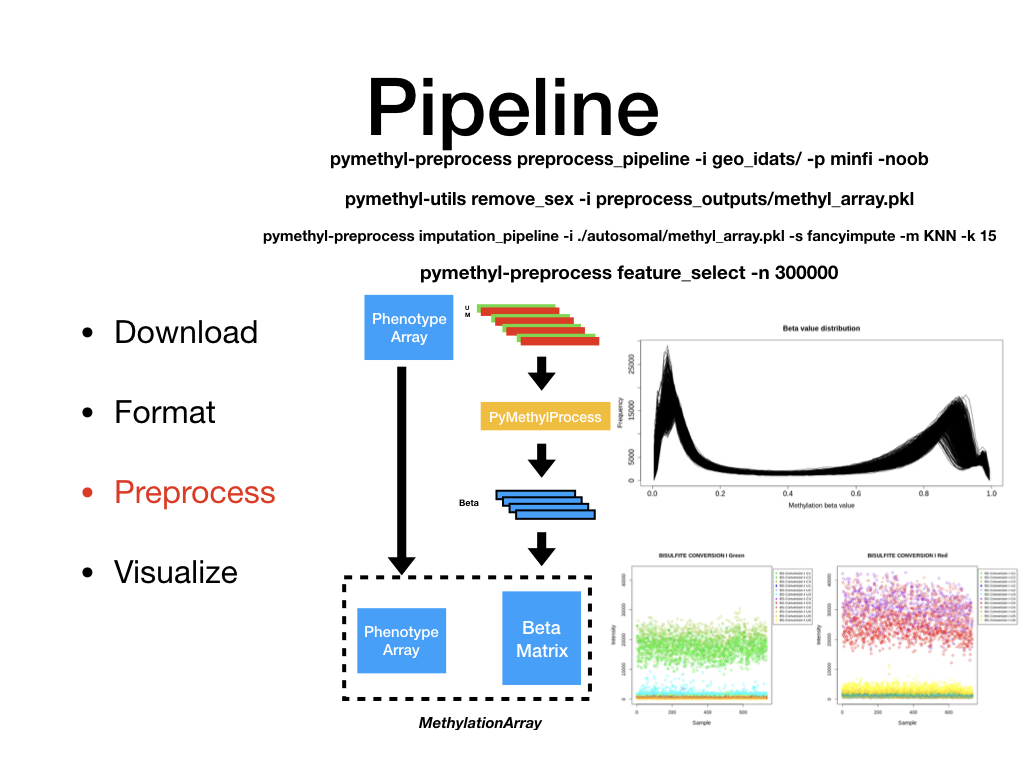
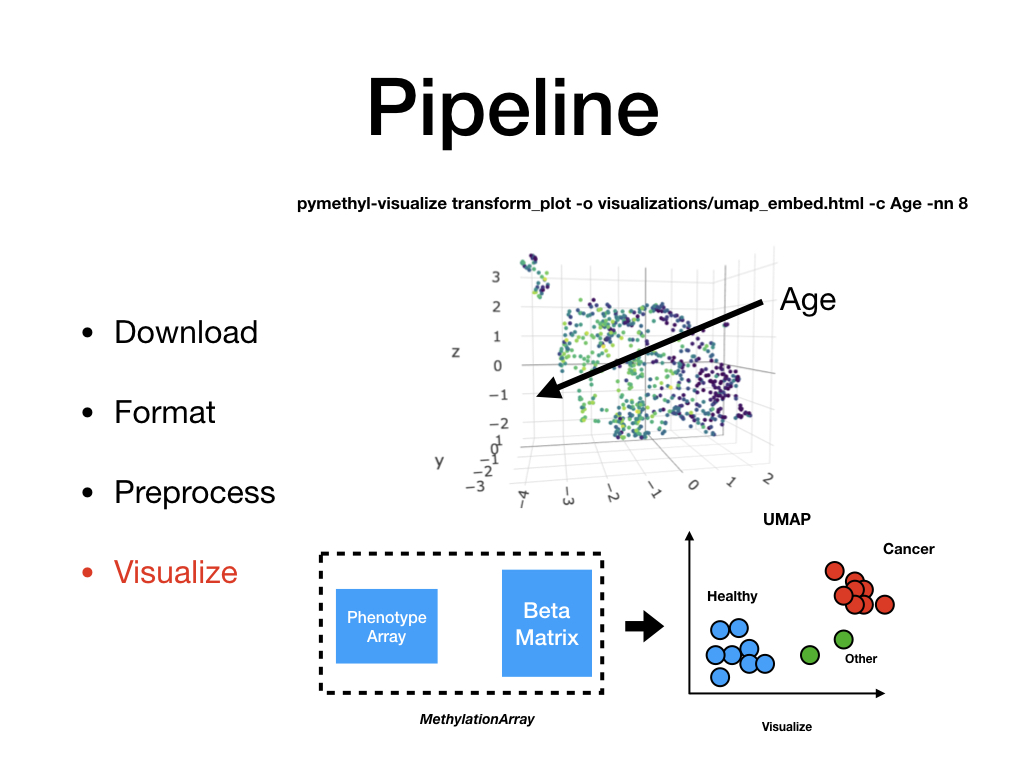
PreProcessDataTypes.py¶
Contains datatypes core to downloading IDATs, preprocessing IDATs and samplesheets.
-
class
pymethylprocess.PreProcessDataTypes.PreProcessIDAT(idat_dir, minfi=None, enmix=None, base=None, meffil=None)[source]¶ Class that will preprocess IDATs using R pipelines.
- idat_dir
- Location of idats or samplesheet csv.
- minfi
- Rpy2 importr minfi library, default to None will load through rpy2
- enmix
- Rpy2 importr enmix library, default to None will load through rpy2
- base
- Rpy2 importr base library, default to None will load through rpy2
- meffil
- Rpy2 importr meffil library, default to None will load through rpy2
-
export_csv(output_dir)[source]¶ Export pheno and beta dataframes to CSVs
- output_dir
- Where to store csvs.
-
export_pickle(output_pickle, disease='')[source]¶ Export pheno and beta dataframes to pickle, stored in python dict that can be loaded into MethylationArray
- output_pickle
- Where to store MethylationArray.
- disease
- Custom naming scheme for data.
-
export_sql(output_db, disease='')[source]¶ Export pheno and beta dataframes to SQL
- output_db
- Where to store data, sqlite db.
- disease
- Custom naming scheme for data.
-
extract_pheno_data(methylset=False)[source]¶ Extract pheno data from MSet or RGSet, minfi.
- methylset
- If MSet has beenn created, set to True, else extract from original RGSet.
-
output_pheno_beta(meffil=False)[source]¶ Get pheno and beta dataframe objects stored as attributes for input to MethylationArray object.
- meffil
- True if ran meffil pipeline.
-
plot_original_qc(output_dir)[source]¶ Plot QC results from ENmix pipeline and possible minfi. Still experimental.
- output_dir
- Where to store plots.
-
plot_qc_metrics(output_dir)[source]¶ Plot QC results from ENmix pipeline and possible minfi. Still experimental.
- output_dir
- Where to store plots.
-
preprocessENmix(n_cores=6)[source]¶ Run ENmix preprocessing pipeline.
- n_cores
- Number of CPUs to use.
-
preprocessMeffil(n_cores=6, n_pcs=4, qc_report_fname='qc/report.html', normalization_report_fname='norm/report.html', pc_plot_fname='qc/pc_plot.pdf', useCache=True, qc_only=True, qc_parameters={'p.beadnum.cpgs': 0.1, 'p.beadnum.samples': 0.1, 'p.detection.cpgs': 0.1, 'p.detection.samples': 0.1}, rm_sex=False)[source]¶ Run meffil preprocessing pipeline with functional normalization.
- n_cores
- Number of CPUs to use.
- n_pcs
- Number of principal components to use for functional normalization, set to -1 to autoselect via kneedle algorithm.
- qc_report_fname
- HTML filename to store QC report.
- normalization_report_fname
- HTML filename to store normalization report
- pc_plot_fname
- PDF file to store principal components plot.
- useCache
- Use saved QC objects instead of running through QC again.
- qc_only
- Perform QC, then save and quit before normalization.
- qc_parameters
- Python dictionary with parameters for qc.
- rm_sex
- Remove non-autosomal cpgs?
-
preprocess_enmix_pipeline(n_cores=6, pipeline='enmix', noob=False, qc_only=False, use_cache=False)[source]¶ Run complete ENmix or minfi preprocessing pipeline.
- n_cores
- Number CPUs.
- pipeline
- Run enmix or minfi
- noob
- Noob norm or RAW if minfi running.
- qc_only
- Save and quit after only running QC?
- use_cache
- Load preexisting RGSet instead of running QC again.
-
class
pymethylprocess.PreProcessDataTypes.PreProcessPhenoData(pheno_sheet, idat_dir, header_line=0)[source]¶ Class that will manipute phenotype samplesheet before preprocessing of IDATs.
- pheno_sheet
- Location of clinical info csv.
- idat_dir
- Location of idats
- header_line
- Where to start reading clinical csv
-
concat(other_formatted_sheet)[source]¶ Concat multiple PreProcessPhenoData objects, concat their dataframes to accept more than one smaplesheet/dataset.
- other_formatted_sheet
- Other PreProcessPhenoData to concat.
-
export(output_sheet_name)[source]¶ Export pheno data to csv after done with manipulation.
- output_sheet_name
- Output csv name.
-
format_custom(basename_col, disease_class_column, include_columns={})[source]¶ Custom format clinical sheet if user supplied idats.
- basename_col
- Column name of sample names.
- disease_class_column
- Disease column of clinical info csv.
- include_columns
- Dictionary specifying other columns to include, and new names to assign them to.
-
format_geo(disease_class_column='methylation class:ch1', include_columns={})[source]¶ Format clinical sheets if downloaded geo idats.
- disease_class_column
- Disease column of clinical info csv.
- include_columns
- Dictionary specifying other columns to include, and new names to assign them to.
-
format_tcga(mapping_file='idat_filename_case.txt')[source]¶ Format clinical sheets if downloaded tcga idats.
- mapping_file
- Maps uuids to proper tcga sample names, should be downloaded with tcga clinical information.
-
get_categorical_distribution(key, disease_only=False, subtype_delimiter=', ')[source]¶ Print categorical distribution, counts for each unique value in phenotype column.
- key
- Phenotype Column.
- disease_only
- Whether to split phenotype column entries by delimiter.
- subtype_delimiter
- Subtype delimiter to split on.
-
merge(other_formatted_sheet, use_second_sheet_disease=True, no_disease_merge=False)[source]¶ Merge multiple PreProcessPhenoData objects, merge their dataframes to accept more than one saplesheet/dataset or add more pheno info.
- other_formatted_sheet
- Other PreProcessPhenoData to merge.
- use_second_sheet_disease
- Change disease column to that of second sheet instead of first.
- no_disease_merge
- Keep both disease columns from both sheets.
-
remove_diseases(exclude_disease_list, low_count, disease_only, subtype_delimiter)[source]¶ Remove samples with certain diseases from disease column.
- exclude_disease_list
- List containing diseases to remove.
- low_count
- Remove samples that have less than x disease occurances in column.
- disease_only
- Whether to split phenotype column entries by delimiter.
- subtype_delimiter
- Subtype delimiter to split on.
-
class
pymethylprocess.PreProcessDataTypes.TCGADownloader[source]¶ Downloads TCGA and GEO IDAT and clinical data
-
download_clinical(output_dir)[source]¶ Download TCGA Clinical Data.
- output_dir
- Where to output clinical data csv.
-
MethylationDataTypes.py¶
Contains datatypes core to storing beta and phenotype methylation data, and imputation.
-
class
pymethylprocess.MethylationDataTypes.ImputerObject(solver, method, opts={})[source]¶ Class that stores and accesses different types of imputers. Construct sklearn-like imputer given certain input arguments.
- solver
- Library for imputation, eg. sklearn, fancyimpute.
- method
- Imputation method in library, named.
- opts
- Additional options to assign to imputer.
-
class
pymethylprocess.MethylationDataTypes.MethylationArray(pheno_df, beta_df, name='')[source]¶ Stores beta and phenotype information and performs various operations. Initialize MethylationArray object by inputting dataframe of phenotypes and dataframe of beta values with samples as index.
- pheno_df
- Phenotype dataframe (samples x covariates)
- beta_df
- Beta Values Dataframe (samples x cpgs)
-
bin_column(col, n_bins)[source]¶ Turn continuous variable/covariate into categorical bins. Returns name of new column and updates phenotype matrix to reflect this change.
- col
- Continuous column of phenotype array to bin.
- n_bins
- Number of bins to create.
-
categorical_breakdown(key)[source]¶ Print categorical distribution, counts for each unique value in phenotype column.
- key
- Phenotype Column.
-
feature_select(n_top_cpgs, feature_selection_method='mad', metric='correlation', nn=10)[source]¶ Perform unsupervised feature selection on MethylationArray.
- n_top_cpgs
- Number of CpGs to retain.
- feature_selection_method
- Method to perform selection.
- metric
- If considering structural feature selection like SPEC, use this distance metric.
- nn
- Number of nearest neighbors.
-
classmethod
from_pickle(input_pickle)[source]¶ Load MethylationArray stored in pickle.
Usage: MethylationArray.from_pickle([input_pickle])
- input_pickle
- Stored MethylationArray pickle.
-
groupby(key)[source]¶ Groupby for Methylation Array. Returns generator of methylation arrays grouped by key.
- preprocess_sample_df
- New phenotype dataframe.
-
impute(imputer)[source]¶ Perform imputation on NaN beta vaues. Input imputater returned from ImputerObject.
- imputer
- Type of imputer object, in sklearn type interface.
-
merge_preprocess_sheet(preprocess_sample_df)[source]¶ Feed in another phenotype dataframe that will be merged with existing phenotype array.
- preprocess_sample_df
- New phenotype dataframe.
-
overwrite_pheno_data(preprocess_sample_df)[source]¶ Feed in another phenotype dataframe that will overwrite overlapping keys of existing phenotype array.
- preprocess_sample_df
- New phenotype dataframe.
-
remove_missingness(cpg_threshold=None, sample_threshold=None)[source]¶ Remove samples and CpGs with certain level of missingness..
- cpg_threshold
- If more than fraction of Samples for this CpG are missing, remove cpg.
- sample_threshold
- If more than fraction of CpGs for this sample are missing, remove sample.
-
remove_na_samples(outcome_cols)[source]¶ Remove samples of MethylationArray who have missing values in phenotype column.
- outcome_cols
- Phenotype columns, if any rows contain missing values, samples are removed.
-
split_by_subtype(disease_only, subtype_delimiter)[source]¶ Split MethylationArray into generator of MethylationArrays by phenotype column. Much akin to groupby. Only splits from disease column.
- disease_only
- Consider disease superclass.
- subtype_delimiter
- How to break up disease column if using disease_only.
-
split_key(key, subtype_delimiter)[source]¶ Manipulate an entire phenotype column, splitting each element up by some delimiter.
- key
- Phenotype column.
- subtype_delimiter
- How to break up strings in columns. S1,s2 -> S1 for instance.
-
split_train_test(train_p=0.8, stratified=True, disease_only=False, key='disease', subtype_delimiter=', ', val_p=0.0)[source]¶ Split MethylationArray into training and test sets, with option to stratify by categorical covariate.
- train_p
- Fraction of methylation array to use as training set.
- stratified
- Whether to stratify by categorical variable.
- disease_only
- Consider disease superclass by some delimiter. For instance if disease is S1,s2, superclass would be S1.
- key
- Column to stratify on.
- subtype_delimiter
- How to split disease column into super/subclass.
- val_p
- If set greater than 0, will create additional validation set, fraction of which is broken off from training set.
-
subsample(key='disease', n_samples=None, frac=None, categorical=False)[source]¶ Subsample MethylationArray, make the set randomly smaller.
- key
- If stratifying, use this column of pheno array.
- n_samples
- Number of samples to consider overall, or per stratum.
- frac
- Alternative to n_samples, where x frac of array or stratum is considered.
- categorical
- Whether to stratify by column.
-
subset_cpgs(cpgs)[source]¶ Subset beta matrix by list of Cpgs. Parameters ———- cpgs
Cpgs to subset by.
-
write_csvs(output_dir)[source]¶ Write phenotype data and beta values to csvs.
- output_dir
- Directory to output csv files.
-
class
pymethylprocess.MethylationDataTypes.MethylationArrays(list_methylation_arrays)[source]¶ Literally a list of methylation arrays, with methods operate on these arrays that is memory efficient. Initialize with list of methylation arrays. Can optionally leave list empty or with one element.
- list_methylation_arrays
- List of methylation arrays.
-
combine(array_generator=None)[source]¶ Combine the list of methylation arrays into one array via concatenation of beta matrices and phenotype arrays.
- array_generator
- Generator of additional methylation arrays for computational memory minimization.
-
pymethylprocess.MethylationDataTypes.extract_pheno_beta_df_from_folder(folder)[source]¶ Return phenotype and beta dataframes from specified folder with csv.
- folder
- Input folder.
meffil_functions.py¶
Contains a few R functions that interact with meffil and minfi.
-
pymethylprocess.meffil_functions.est_cell_counts_IDOL(rgset, library)[source]¶ Given RGSet object, estimate cell counts for 450k/850k using reference approach via IDOL library.
- rgset
- RGSet object stored in python via rpy2
- library
- What type of CpG library to use.
-
pymethylprocess.meffil_functions.est_cell_counts_meffil(qc_list, cell_type_reference)[source]¶ Given QCObject list R object, estimate cell counts using reference approach via meffil.
- qc_list
- R list containing qc objects.
- cell_type_reference
- Reference blood/tissue set.
-
pymethylprocess.meffil_functions.est_cell_counts_minfi(rgset)[source]¶ Given RGSet object, estimate cell counts using reference approach via minfi.
- rgset
- RGSet object stored in python via rpy2
-
pymethylprocess.meffil_functions.load_detection_p_values_beadnum(qc_list, n_cores)[source]¶ Return list of detection p-value matrix and bead number matrix.
- qc_list
- R list containing qc objects.
- n_cores
- Number of cores to use in computation.
-
pymethylprocess.meffil_functions.r_autosomal_cpgs(array_type='450k')[source]¶ Return list of autosomal cpg probes per platform.
- array_type
- 450k/850k array?
-
pymethylprocess.meffil_functions.r_snp_cpgs(array_type='450k')[source]¶ Return list of SNP cpg probes per platform.
- array_type
- 450k/850k array?
-
pymethylprocess.meffil_functions.remove_sex(beta, array_type='450k')[source]¶ Remove non-autosomal cpgs from beta matrix.
- array_type
- 450k/850k array?
-
pymethylprocess.meffil_functions.set_missing(beta, pval_beadnum, detection_val=1e-06)[source]¶ Set missing beta values to NA, taking into account detection values and bead number thesholds.
- pval_beadnum
- Detection pvalues and number of beads per cpg/samples
- detection_val
- If threshold to set site to missingness based on p-value detection.
general_machine_learning.py¶
Contains a machine learning class to perform scikit-learn like operations, along with held-out hyperparameter grid search.
-
class
pymethylprocess.general_machine_learning.MachineLearning(model, options, grid={}, labelencode=False, n_eval=0)[source]¶ Machine learning class to run sklearn-like pipeline on MethylationArray data. Initialize object with scikit-learn model, and optionally supply a hyperparameter search grid.
- model
- Scikit-learn-like model, classification, regression, dimensionality reduction, clustering etc.
- options
- Options to supply model in form of dictionary.
- grid
- Alternatively, supply search grid to search for bets hyperparameters.
- labelencode
- T/F encode string labels.
- n_eval
- Number of evaluations for randomized grid search, if set to 0, perform exhaustive grid search
-
assign_results_to_pheno_col(methyl_array, new_col, output_pkl)[source]¶ Assign results to new phenotype column.
- methyl_array
- MethylationArray.
- new_col
- New column name.
- output_pkl
- Output pickle to dump MethylationArray to.
-
fit(train_methyl_array, val_methyl_array=None, outcome_cols=None)[source]¶ Fit data to model.
- train_methyl_array
- Training MethylationArray.
- val_methyl_array
- Validation MethylationArray. Can set to None.
- outcome_cols
- Set to none if not needed, but phenotype column to train on, can be multiple.
-
fit_predict(train_methyl_array, outcome_cols=None)[source]¶ Fit and predict training data.
- train_methyl_array
- Training MethylationArray.
- outcome_cols
- Set to none if not needed, but phenotype column to train on, can be multiple.
-
fit_transform(train_methyl_array, outcome_cols=None)[source]¶ Fit and transform to training data.
- train_methyl_array
- Training MethylationArray.
- outcome_cols
- Set to none if not needed, but phenotype column to train on, can be multiple.
-
predict(test_methyl_array)[source]¶ Make new predictions on test methylation array.
- test_methyl_array
- Testing MethylationArray.
-
return_outcome_metric(methyl_array, outcome_cols, metric, run_bootstrap=False)[source]¶ Supply metric to evaluate results.
- methyl_array
- MethylationArray to evaluate.
- outcome_cols
- Outcome phenotype columns.
- metric
- Sklearn evaluation metric.
- run_bootstrap
- Make 95% CI from 1k bootstraps.
-
store_results(output_pkl, results_dict={})[source]¶ Store results in pickle file.
- output_pkl
- Output pickle to dump results to.
- results_dict
- Supply own results dict to be dumped.
pymethyl-install¶
pymethyl-install [OPTIONS] COMMAND [ARGS]...
Options
-
--version¶ Show the version and exit.
change_gcc_path¶
Change GCC and G++ paths if don’t have version 7.2.0. [Experimental]
pymethyl-install change_gcc_path [OPTIONS]
install_custom¶
Installs bioconductor packages.
pymethyl-install install_custom [OPTIONS]
Options
-
-p,--package<package>¶ Custom packages. [default: ENmix]
-
-m,--manager¶ Use BiocManager (recommended).
install_minfi_others¶
Installs minfi and other dependencies.
pymethyl-install install_minfi_others [OPTIONS]
install_r_packages¶
Installs r packages.
pymethyl-install install_r_packages [OPTIONS]
Options
-
-p,--package<package>¶ Custom packages. [default: ]
install_some_deps¶
Installs bioconductor, minfi, enmix, tcga biolinks, and meffil.
pymethyl-install install_some_deps [OPTIONS]
pymethyl-visualize¶
pymethyl-visualize [OPTIONS] COMMAND [ARGS]...
Options
-
--version¶ Show the version and exit.
plot_cell_type_results¶
Plot csv containing cell type results into side by side boxplots.
pymethyl-visualize plot_cell_type_results [OPTIONS]
Options
-
-i,--input_csv<input_csv>¶ Input csv. [default: cell_type_estimates.csv]
-
-o,--outfilename<outfilename>¶ Output png. [default: visualizations/cell_type_results.png]
-
-cols,--plot_cols<plot_cols>¶ Plot columns. [default: Gran, CD4T, CD8T, Bcell, Mono, NK, gMDSC]
-
-fs,--font_scale<font_scale>¶ Font scaling [default: 1.0]
plot_heatmap¶
Plot heatmap from CSV file.
pymethyl-visualize plot_heatmap [OPTIONS]
Options
-
-i,--input_csv<input_csv>¶ Input csv. [default: ]
-
-o,--outfilename<outfilename>¶ Output png. [default: output.png]
-
-idx,--index_col<index_col>¶ Index load dataframe [default: 0]
-
-fs,--font_scale<font_scale>¶ Font scaling [default: 1.0]
-
-min,--min_val<min_val>¶ Min heat val [default: 0.0]
-
-max,--max_val<max_val>¶ Max heat val, if -1, defaults to None [default: 1.0]
-
-a,--annot¶ Annotate heatmap [default: False]
-
-n,--norm¶ Normalize matrix data [default: False]
-
-c,--cluster¶ Cluster matrix data [default: False]
-
-m,--matrix_type<matrix_type>¶ Type of matrix supplied [default: none]
-
-x,--xticks¶ Show x ticks [default: False]
-
-y,--yticks¶ Show y ticks [default: False]
-
-t,--transpose¶ Transpose matrix data [default: False]
-
-col,--color_column<color_column>¶ Color column. [default: color]
transform_plot¶
Dimensionality reduce VAE or original beta values using UMAP and plot using plotly.
pymethyl-visualize transform_plot [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
-
-c,--column_of_interest<column_of_interest>¶ Column extract from phenotype data. [default: disease]
-
-o,--output_file<output_file>¶ Output visualization. [default: ./visualization.html]
-
-nn,--n_neighbors<n_neighbors>¶ Number of neighbors UMAP. [default: 5]
-
-a,--axes_off¶ Whether to turn axes on or off.
-
-s,--supervised¶ Supervise umap embedding.
-
-d,--min_dist<min_dist>¶ UMAP min distance. [default: 0.1]
-
-m,--metric<metric>¶ Reduction metric. [default: euclidean]
-
-cc,--case_control_override¶ Add controls from case_control column and override current disease for classification tasks. [default: False]
pymethyl-preprocess¶
pymethyl-preprocess [OPTIONS] COMMAND [ARGS]...
Options
-
--version¶ Show the version and exit.
batch_deploy_preprocess¶
Deploy multiple preprocessing jobs in series or parallel.
pymethyl-preprocess batch_deploy_preprocess [OPTIONS]
Options
-
-n,--n_cores<n_cores>¶ Number cores to use for preprocessing. [default: 6]
-
-i,--subtype_output_dir<subtype_output_dir>¶ Output subtypes pheno csv. [default: ./preprocess_outputs/]
-
-m,--meffil¶ Preprocess using meffil.
-
-t,--torque¶ Job submission torque.
-
-r,--run¶ Actually run local job or just print out command.
-
-s,--series¶ Run commands in series.
-
-p,--pc_qc_parameters_csv<pc_qc_parameters_csv>¶ For meffil, qc parameters and pcs for final qc and functional normalization. [default: ./preprocess_outputs/pc_qc_parameters.csv]
-
-u,--use_cache¶ If this is selected, loads qc results rather than running qc again. Only works for meffil selection.
-
-qc,--qc_only¶ Only perform QC for meffil pipeline, caches results into rds file for loading again, only works if use_cache is false.
-
-c,--chunk_size<chunk_size>¶ If not series, chunk up and run these number of commands at once.. -1 means all commands at once.
combine_methylation_arrays¶
If split MethylationArrays by subtype for either preprocessing or imputation, can use to recombine data for downstream step.
pymethyl-preprocess combine_methylation_arrays [OPTIONS]
Options
-
-i,--input_pkls<input_pkls>¶ Input pickles for beta and phenotype data. [default: ./preprocess_outputs/methyl_array.pkl]
-
-d,--optional_input_pkl_dir<optional_input_pkl_dir>¶ Auto grab input pkls. [default: ]
-
-o,--output_pkl<output_pkl>¶ Output database for beta and phenotype data. [default: ./combined_outputs/methyl_array.pkl]
-
-e,--exclude<exclude>¶ If -d selected, these diseases will be excluded from study. [default: ]
concat_sample_sheets¶
Concat two sample files for more fields for minfi+ input, adds more samples.
pymethyl-preprocess concat_sample_sheets [OPTIONS]
Options
-
-s1,--sample_sheet1<sample_sheet1>¶ Clinical information downloaded from tcga/geo/custom, formatted using create_sample_sheet. [default: ./tcga_idats/clinical_info1.csv]
-
-s2,--sample_sheet2<sample_sheet2>¶ Clinical information downloaded from tcga/geo/custom, formatted using create_sample_sheet. [default: ./tcga_idats/clinical_info2.csv]
-
-os,--output_sample_sheet<output_sample_sheet>¶ CSV for minfi input. [default: ./tcga_idats/minfiSheet.csv]
create_sample_sheet¶
Create sample sheet for input to minfi, meffil, or enmix.
pymethyl-preprocess create_sample_sheet [OPTIONS]
Options
-
-is,--input_sample_sheet<input_sample_sheet>¶ Clinical information downloaded from tcga/geo/custom. [default: ./tcga_idats/clinical_info.csv]
-
-s,--source_type<source_type>¶ Source type of data. [default: tcga]
-
-i,--idat_dir<idat_dir>¶ Idat directory. [default: ./tcga_idats/]
-
-os,--output_sample_sheet<output_sample_sheet>¶ CSV for minfi input. [default: ./tcga_idats/minfiSheet.csv]
-
-m,--mapping_file<mapping_file>¶ Mapping file from uuid to TCGA barcode. Downloaded using download_tcga. [default: ./idat_filename_case.txt]
-
-l,--header_line<header_line>¶ Line to begin reading csv/xlsx. [default: 0]
-
-d,--disease_class_column<disease_class_column>¶ Disease classification column, for custom and geo datasets. [default: methylation class:ch1]
-
-b,--basename_col<basename_col>¶ Basename classification column, for custom datasets. [default: Sentrix ID (.idat)]
-
-c,--include_columns_file<include_columns_file>¶ Custom columns file containing columns to keep, separated by n. Add a tab for each line if you wish to rename columns: original_name t new_column_name [default: ]
download_clinical¶
Download all TCGA 450k clinical info.
pymethyl-preprocess download_clinical [OPTIONS]
Options
-
-o,--output_dir<output_dir>¶ Output directory for exported idats. [default: ./tcga_idats/]
download_geo¶
Download geo methylation study idats and clinical info.
pymethyl-preprocess download_geo [OPTIONS]
Options
-
-g,--geo_query<geo_query>¶ GEO study to query. [default: ]
-
-o,--output_dir<output_dir>¶ Output directory for exported idats. [default: ./geo_idats/]
download_tcga¶
Download all tcga 450k data.
pymethyl-preprocess download_tcga [OPTIONS]
Options
-
-o,--output_dir<output_dir>¶ Output directory for exported idats. [default: ./tcga_idats/]
feature_select¶
Filter CpGs by taking x top CpGs with highest mean absolute deviation scores or via spectral feature selection.
pymethyl-preprocess feature_select [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./imputed_outputs/methyl_array.pkl]
-
-o,--output_pkl<output_pkl>¶ Output database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
-
-n,--n_top_cpgs<n_top_cpgs>¶ Number cpgs to include with highest variance across population. [default: 300000]
-
-f,--feature_selection_method<feature_selection_method>¶
-
-mm,--metric<metric>¶
-
-nn,--n_neighbors<n_neighbors>¶ Number neighbors for feature selection, default enacts rbf kernel. [default: 0]
-
-m,--mad_top_cpgs<mad_top_cpgs>¶ Number cpgs to apply mad filtering first before more sophisticated feature selection. If 0 or primary feature selection is mad, no mad pre-filtering. [default: 0]
get_categorical_distribution¶
Get categorical distribution of columns of sample sheet.
pymethyl-preprocess get_categorical_distribution [OPTIONS]
Options
-
-is,--formatted_sample_sheet<formatted_sample_sheet>¶ Clinical information downloaded from tcga/geo/custom, formatted using create_sample_sheet. [default: ./tcga_idats/minfiSheet.csv]
-
-k,--key<key>¶ Column of csv to print statistics for. [default: disease]
-
-d,--disease_only¶ Only look at disease, or text before subtype_delimiter.
-
-sd,--subtype_delimiter<subtype_delimiter>¶ Delimiter for disease extraction. [default: ,]
imputation_pipeline¶
Imputation of subtype or no subtype using various imputation methods.
pymethyl-preprocess imputation_pipeline [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./combined_outputs/methyl_array.pkl]
-
-ss,--split_by_subtype¶ Imputes CpGs by subtype before combining again.
-
-m,--method<method>¶ Method of imputation. [default: KNN]
-
-s,--solver<solver>¶ Imputation library. [default: fancyimpute]
-
-k,--n_neighbors<n_neighbors>¶ Number neighbors for imputation if using KNN. [default: 5]
-
-r,--orientation<orientation>¶ Impute CpGs or samples. [default: Samples]
-
-o,--output_pkl<output_pkl>¶ Output database for beta and phenotype data. [default: ./imputed_outputs/methyl_array.pkl]
-
-n,--n_top_cpgs<n_top_cpgs>¶ Number cpgs to include with highest variance across population. Greater than 0 allows for mad filtering during imputation to skip mad step. [default: 0]
-
-f,--feature_selection_method<feature_selection_method>¶
-
-mm,--metric<metric>¶
-
-nfs,--n_neighbors_fs<n_neighbors_fs>¶ Number neighbors for feature selection, default enacts rbf kernel. [default: 0]
-
-d,--disease_only¶ Only look at disease, or text before subtype_delimiter.
-
-sd,--subtype_delimiter<subtype_delimiter>¶ Delimiter for disease extraction. [default: ,]
-
-st,--sample_threshold<sample_threshold>¶ Value between 0 and 1 for NaN removal. If samples has sample_threshold proportion of cpgs missing, then remove sample. Set to -1 to not remove samples. [default: -1.0]
-
-ct,--cpg_threshold<cpg_threshold>¶ Value between 0 and 1 for NaN removal. If cpgs has cpg_threshold proportion of samples missing, then remove cpg. Set to -1 to not remove samples. [default: -1.0]
meffil_encode¶
Reformat file for meffil input.
pymethyl-preprocess meffil_encode [OPTIONS]
Options
-
-is,--input_sample_sheet<input_sample_sheet>¶ CSV for minfi input. [default: ./tcga_idats/minfiSheet.csv]
-
-os,--output_sample_sheet<output_sample_sheet>¶ CSV for minfi input. [default: ./tcga_idats/minfiSheet.csv]
merge_sample_sheets¶
Merge two sample files for more fields for minfi+ input.
pymethyl-preprocess merge_sample_sheets [OPTIONS]
Options
-
-s1,--sample_sheet1<sample_sheet1>¶ Clinical information downloaded from tcga/geo/custom, formatted using create_sample_sheet. [default: ./tcga_idats/clinical_info1.csv]
-
-s2,--sample_sheet2<sample_sheet2>¶ Clinical information downloaded from tcga/geo/custom, formatted using create_sample_sheet. [default: ./tcga_idats/clinical_info2.csv]
-
-os,--output_sample_sheet<output_sample_sheet>¶ CSV for minfi input. [default: ./tcga_idats/minfiSheet.csv]
-
-d,--second_sheet_disease¶ Use second sheet’s disease column.
-
-nd,--no_disease_merge¶ Don’t merge disease columns.
na_report¶
Print proportion of missing values throughout dataset.
pymethyl-preprocess na_report [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./preprocess_outputs/methyl_array.pkl]
-
-o,--output_dir<output_dir>¶ Output database for na report. [default: ./na_report/]
-
-r,--head_directory¶ -i option becomes directory, and searches there for multiple input pickles.
preprocess_pipeline¶
Perform preprocessing of idats using enmix or meffil.
pymethyl-preprocess preprocess_pipeline [OPTIONS]
Options
-
-i,--idat_dir<idat_dir>¶ Idat dir for one sample sheet, alternatively can be your phenotype sample sheet. [default: ./tcga_idats/]
-
-n,--n_cores<n_cores>¶ Number cores to use for preprocessing. [default: 6]
-
-o,--output_pkl<output_pkl>¶ Output database for beta and phenotype data. [default: ./preprocess_outputs/methyl_array.pkl]
-
-m,--meffil¶ Preprocess using meffil.
-
-pc,--n_pcs<n_pcs>¶ For meffil, number of principal components for functional normalization. If set to -1, then PCs are selected using elbow method. [default: -1]
-
-p,--pipeline<pipeline>¶ If not meffil, preprocess using minfi or enmix. [default: enmix]
-
-noob,--noob_norm¶ Run noob normalization of minfi selected.
-
-u,--use_cache¶ If this is selected, loads qc results rather than running qc again and update with new qc parameters. Only works for meffil selection. Minfi and enmix just loads RG Set.
-
-qc,--qc_only¶ Only perform QC for meffil pipeline, caches results into rds file for loading again, only works if use_cache is false. Minfi and enmix just saves the RGSet before preprocessing.
-
-bns,--p_beadnum_samples<p_beadnum_samples>¶ From meffil documentation, “fraction of probes that failed the threshold of 3 beads”. [default: 0.05]
-
-pds,--p_detection_samples<p_detection_samples>¶ From meffil documentation, “fraction of probes that failed a detection.pvalue threshold of 0.01”. [default: 0.05]
-
-bnc,--p_beadnum_cpgs<p_beadnum_cpgs>¶ From meffil documentation, “fraction of samples that failed the threshold of 3 beads”. [default: 0.05]
-
-pdc,--p_detection_cpgs<p_detection_cpgs>¶ From meffil documentation, “fraction of samples that failed a detection.pvalue threshold of 0.01”. [default: 0.05]
-
-sc,--sex_cutoff<sex_cutoff>¶ From meffil documentation, “difference of total median intensity for Y chromosome probes and X chromosome probes”. [default: -2]
-
-sd,--sex_sd<sex_sd>¶ From meffil documentation, “sex detection outliers if outside this range”. [default: 5]
remove_diseases¶
Exclude diseases from study by count number or exclusion list.
pymethyl-preprocess remove_diseases [OPTIONS]
Options
-
-is,--formatted_sample_sheet<formatted_sample_sheet>¶ Clinical information downloaded from tcga/geo/custom, formatted using create_sample_sheet. [default: ./tcga_idats/clinical_info.csv]
-
-e,--exclude_disease_list<exclude_disease_list>¶ List of conditions to exclude, from disease column, comma delimited. [default: ]
-
-os,--output_sheet_name<output_sheet_name>¶ CSV for minfi input. [default: ./tcga_idats/minfiSheet.csv]
-
-l,--low_count<low_count>¶ Remove diseases if they are below a certain count, default this is not used. [default: 0]
-
-d,--disease_only¶ Only look at disease, or text before subtype_delimiter.
-
-sd,--subtype_delimiter<subtype_delimiter>¶ Delimiter for disease extraction. [default: ,]
split_preprocess_input_by_subtype¶
Split preprocess input samplesheet by disease subtype.
pymethyl-preprocess split_preprocess_input_by_subtype [OPTIONS]
Options
-
-i,--idat_csv<idat_csv>¶ Idat csv for one sample sheet, alternatively can be your phenotype sample sheet. [default: ./tcga_idats/minfiSheet.csv]
-
-d,--disease_only¶ Only look at disease, or text before subtype_delimiter.
-
-sd,--subtype_delimiter<subtype_delimiter>¶ Delimiter for disease extraction. [default: ,]
-
-o,--subtype_output_dir<subtype_output_dir>¶ Output subtypes pheno csv. [default: ./preprocess_outputs/]
pymethyl-utils¶
pymethyl-utils [OPTIONS] COMMAND [ARGS]...
Options
-
--version¶ Show the version and exit.
backup_pkl¶
Copy methylarray pickle to new location to backup.
pymethyl-utils backup_pkl [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
-
-o,--output_pkl<output_pkl>¶ Output database for beta and phenotype data. [default: ./backup/methyl_array.pkl]
bin_column¶
Convert continuous phenotype column into categorical by binning.
pymethyl-utils bin_column [OPTIONS]
Options
-
-t,--test_pkl<test_pkl>¶ Pickle containing testing set. [default: ./train_val_test_sets/test_methyl_array.pkl]
-
-c,--col<col>¶ Column to turn into bins. [default: age]
-
-n,--n_bins<n_bins>¶ Number of bins. [default: 10]
-
-ot,--output_test_pkl<output_test_pkl>¶ Binned shap pickle for further testing. [default: ./train_val_test_sets/test_methyl_array_shap_binned.pkl]
concat_csv¶
Concatenate two csv files together.
pymethyl-utils concat_csv [OPTIONS]
Options
-
-i1,--input_csv<input_csv>¶ Beta csv. [default: ./beta1.csv]
-
-i2,--input_csv2<input_csv2>¶ Beta/other csv 2. [default: ./cell_estimates.csv]
-
-o,--output_csv<output_csv>¶ Output csv. [default: ./beta.concat.csv]
-
-a,--axis<axis>¶ Axis to merge on. Columns are 0, rows are 1. [default: 1]
counts¶
Return categorical breakdown of phenotype column.
pymethyl-utils counts [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
-
-k,--key<key>¶ Key to split on. [default: disease]
create_external_validation_set¶
Create external validation set containing same CpGs as training set.
pymethyl-utils create_external_validation_set [OPTIONS]
Options
-
-t,--train_pkl<train_pkl>¶ Input methyl array. [default: ./train_val_test_sets/train_methyl_array.pkl]
-
-q,--query_pkl<query_pkl>¶ Input methylation array to add/subtract cpgs to. [default: ./final_preprocessed/methyl_array.pkl]
-
-o,--output_pkl<output_pkl>¶ Output methyl array external validation. [default: ./external_validation/methyl_array.pkl]
-
-c,--cpg_replace_method<cpg_replace_method>¶ What to do for missing CpGs. [default: mid]
feature_select_train_val_test¶
Filter CpGs by taking x top CpGs with highest mean absolute deviation scores or via spectral feature selection.
pymethyl-utils feature_select_train_val_test [OPTIONS]
Options
-
-i,--input_pkl_dir<input_pkl_dir>¶ Input database for beta and phenotype data. [default: ./train_val_test_sets/]
-
-o,--output_dir<output_dir>¶ Output database for beta and phenotype data. [default: ./train_val_test_sets_fs/]
-
-n,--n_top_cpgs<n_top_cpgs>¶ Number cpgs to include with highest variance across population. [default: 300000]
-
-f,--feature_selection_method<feature_selection_method>¶
-
-mm,--metric<metric>¶
-
-nn,--n_neighbors<n_neighbors>¶ Number neighbors for feature selection, default enacts rbf kernel. [default: 0]
-
-m,--mad_top_cpgs<mad_top_cpgs>¶ Number cpgs to apply mad filtering first before more sophisticated feature selection. If 0 or primary feature selection is mad, no mad pre-filtering. [default: 0]
fix_key¶
Format certain column of phenotype array in MethylationArray.
pymethyl-utils fix_key [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
-
-k,--key<key>¶ Key to split on. [default: disease]
-
-d,--disease_only¶ Only look at disease, or text before subtype_delimiter.
-
-sd,--subtype_delimiter<subtype_delimiter>¶ Delimiter for disease extraction. [default: ,]
-
-o,--output_pkl<output_pkl>¶ Input database for beta and phenotype data. [default: ./fixed_preprocessed/methyl_array.pkl]
modify_pheno_data¶
Use another spreadsheet to add more descriptive data to methylarray.
pymethyl-utils modify_pheno_data [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
-
-is,--input_formatted_sample_sheet<input_formatted_sample_sheet>¶ Information passed through function create_sample_sheet, has Basename and disease fields. [default: ./tcga_idats/minfi_sheet.csv]
-
-o,--output_pkl<output_pkl>¶ Output database for beta and phenotype data. [default: ./modified_processed/methyl_array.pkl]
move_jpg¶
Move preprocessing jpegs to preprocessing output directory.
pymethyl-utils move_jpg [OPTIONS]
Options
-
-i,--input_dir<input_dir>¶ Directory containing jpg. [default: ./]
-
-o,--output_dir<output_dir>¶ Output directory for images. [default: ./preprocess_output_images/]
overwrite_pheno_data¶
Use another spreadsheet to add more descriptive data to methylarray.
pymethyl-utils overwrite_pheno_data [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
-
-is,--input_formatted_sample_sheet<input_formatted_sample_sheet>¶ Information passed through function create_sample_sheet, has Basename and disease fields. [default: ./tcga_idats/minfi_sheet.csv]
-
-o,--output_pkl<output_pkl>¶ Output database for beta and phenotype data. [default: ./modified_processed/methyl_array.pkl]
-
-c,--index_col<index_col>¶ Index col when reading csv. [default: 0]
pkl_to_csv¶
Output methylarray pickle to csv.
pymethyl-utils pkl_to_csv [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
-
-o,--output_dir<output_dir>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/]
-
-c,--col<col>¶ Column to color. [default: ]
print_number_sex_cpgs¶
Print number of non-autosomal CpGs.
pymethyl-utils print_number_sex_cpgs [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
-
-a,--array_type<array_type>¶ Array Type. [default: 450k]
print_shape¶
Print dimensions of beta matrix.
pymethyl-utils print_shape [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
ref_estimate_cell_counts¶
Reference based cell type estimates.
pymethyl-utils ref_estimate_cell_counts [OPTIONS]
Options
-
-ro,--input_r_object_dir<input_r_object_dir>¶ Input directory containing qc data. [default: ./preprocess_outputs/]
-
-a,--algorithm<algorithm>¶ Algorithm to run cell type. [default: meffil]
-
-ref,--reference<reference>¶ Cell Type Reference. [default: cord blood gse68456]
-
-l,--library<library>¶ IDOL Library. [default: IDOLOptimizedCpGs450klegacy]
-
-o,--output_csv<output_csv>¶ Output cell type estimates. [default: ./added_cell_counts/cell_type_estimates.csv]
remove_sex¶
Remove non-autosomal CpGs.
pymethyl-utils remove_sex [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./preprocess_outputs/methyl_array.pkl]
-
-o,--output_pkl<output_pkl>¶ Output methyl array autosomal. [default: ./autosomal/methyl_array.pkl]
-
-a,--array_type<array_type>¶ Array Type. [default: 450k]
remove_snps¶
Remove SNPs from methylation array.
pymethyl-utils remove_snps [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./autosomal/methyl_array.pkl]
-
-o,--output_pkl<output_pkl>¶ Output methyl array autosomal. [default: ./no_snp/methyl_array.pkl]
-
-a,--array_type<array_type>¶ Array Type. [default: 450k]
set_part_array_background¶
Set subset of CpGs from beta matrix to background values.
pymethyl-utils set_part_array_background [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input methyl array. [default: ./final_preprocessed/methyl_array.pkl]
-
-c,--cpg_pkl<cpg_pkl>¶ Pickled numpy array for subsetting. [default: ./subset_cpgs.pkl]
-
-o,--output_pkl<output_pkl>¶ Output methyl array external validation. [default: ./removal/methyl_array.pkl]
stratify¶
Split methylation array by key and store.
pymethyl-utils stratify [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
-
-k,--key<key>¶ Key to split on. [default: disease]
-
-o,--output_dir<output_dir>¶ Output directory for stratified. [default: ./stratified/]
subset_array¶
Only retain certain number of CpGs from methylation array.
pymethyl-utils subset_array [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input methyl array. [default: ./final_preprocessed/methyl_array.pkl]
-
-c,--cpg_pkl<cpg_pkl>¶ Pickled numpy array for subsetting. [default: ./subset_cpgs.pkl]
-
-o,--output_pkl<output_pkl>¶ Output methyl array external validation. [default: ./subset/methyl_array.pkl]
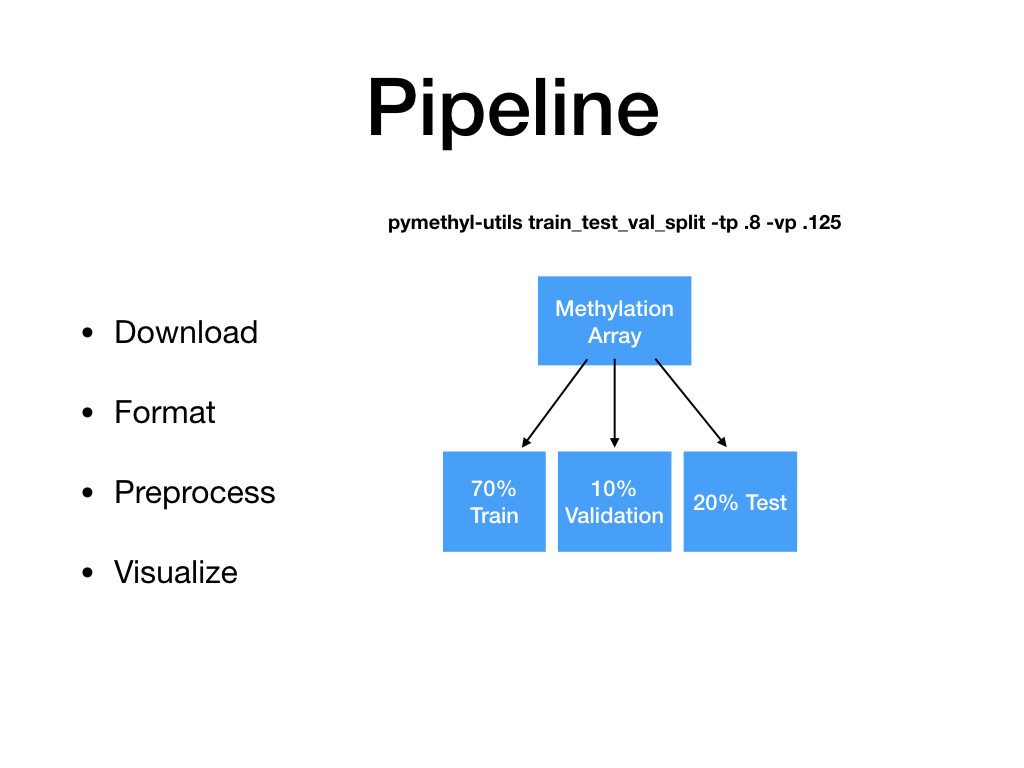
train_test_val_split¶
Split methylation array into train, test, val.
pymethyl-utils train_test_val_split [OPTIONS]
Options
-
-i,--input_pkl<input_pkl>¶ Input database for beta and phenotype data. [default: ./final_preprocessed/methyl_array.pkl]
-
-o,--output_dir<output_dir>¶ Output directory for training, testing, and validation sets. [default: ./train_val_test_sets/]
-
-tp,--train_percent<train_percent>¶ Percent data training on. [default: 0.8]
-
-vp,--val_percent<val_percent>¶ Percent of training data that comprises validation set. [default: 0.1]
-
-cat,--categorical¶ Multi-class prediction. [default: False]
-
-do,--disease_only¶ Only look at disease, or text before subtype_delimiter.
-
-k,--key<key>¶ Key to split on. [default: disease]
-
-sd,--subtype_delimiter<subtype_delimiter>¶ Delimiter for disease extraction. [default: ,]